A lo largo de 12 años de seguimiento, el paciente ha recibido múltiples esquemas terapéuticos que han incluido fármacos antiepilépticos como oxcarbazepina, fenobarbital, ácido valproico, topiramato, levetiracetam y clobazam, siempre en dosis máximas toleradas.

Un niño de 7 años, sin antecedentes médicos relevantes de ningún tipo y con un desarrollo neurológico completamente normal hasta el momento del inicio de los síntomas, acudió a consulta por un episodio febril (período de tiempo en el que la temperatura corporal de una persona está por encima de lo normal), que fue manejado de forma ambulatoria con cefalexina bajo la sospecha de una posible sinusitis, aunque este diagnóstico nunca fue confirmado.
Ocho días después del inicio de la fiebre, el paciente desarrolló un intenso dolor de cabeza que persistió hasta motivar nuevamente la consulta médica.
Aproximadamente 48 horas después del inicio de la cefalea, el niño presentó su primer episodio convulsivo caracterizado por movimientos de versión de la cabeza y los labios hacia el lado izquierdo, seguido de actividad convulsiva clónica generalizada que duraba entre uno y dos minutos.
Estas crisis comenzaron a hacerse cada vez más frecuentes, llevando a un deterioro progresivo del nivel de conciencia hasta evolucionar a un estado epiléptico motor que requirió manejo en unidad de cuidados intensivos.
En la unidad de cuidados intensivos se inició coma barbitúrico (coma inducido) como parte del manejo del estado epiléptico refractario. Durante este período, que se extendió por seis semanas, el paciente continuó presentando crisis convulsivas multifocales de tipo clónico (los músculos se contraen y relajan rítmicamente, creando movimientos sacudidos o temblores) y mioclónico (sacudidas rápidas y breves de los músculos) a pesar del tratamiento intensivo.
Se emplearon múltiples esquemas terapéuticos que incluyeron desde anestésicos intravenosos como tiosental sódico, propofol y midazolam, hasta antiepilépticos convencionales como ácido valproico, fenitoína y clonazepam, además de alternativas terapéuticas como lidocaína intravenosa y topiramato.
En el momento de máxima complejidad del manejo, se llegaron a utilizar simultáneamente seis fármacos antiepilépticos diferentes en sus dosis máximas recomendadas.
Durante la fase aguda se llevó a cabo una exhaustiva evaluación diagnóstica que incluyó estudios para descartar causas infecciosas mediante PCR, cultivos y serologías, así como perfiles metabólicos y toxicológicos completos.
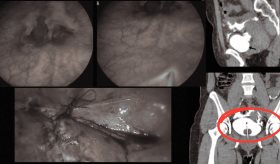
Se realizó además un estudio autoinmune con determinación de anticuerpos neurales y neuroimágenes iniciales que incluyeron resonancia magnética cerebral sin que se encontraran hallazgos patológicos significativos.
El monitoreo electroencefalográfico continuo fue fundamental para caracterizar la actividad epileptiforme.
Después de las seis semanas de tratamiento intensivo, se logró finalmente controlar el estado epiléptico superrefractario. Sin embargo, el paciente evolucionó hacia una epilepsia focal farmacorresistente que se ha mantenido durante los doce años de seguimiento.
Esta condición se ha caracterizado por la presencia de crisis focales con alteración de la conciencia que ocurren entre dos y tres veces por semana, además de eventos mensuales con generalización tónico-clónica.
Estos episodios se han asociado consistentemente con fenómenos autonómicos como bradicardia y desaturación de oxígeno, y han llevado al desarrollo de un déficit cognitivo moderado documentado.
A lo largo de los doce años de seguimiento, el paciente ha recibido múltiples esquemas terapéuticos que han incluido diversas combinaciones de fármacos antiepilépticos como oxcarbazepina, fenobarbital, ácido valproico, topiramato, levetiracetam y clobazam, siempre en dosis máximas toleradas.
Se ha implementado además terapia inmunomoduladora con inmunoglobulina intravenosa durante seis meses consecutivos y dieta cetogénica de bajo índice glicémico. Recientemente se añadió lacosamida al esquema terapéutico basal en un intento por mejorar el control de las crisis.
Hallazgos paraclínicos en fase crónica
Los estudios de seguimiento han mostrado cambios estructurales en el sistema nervioso central, particularmente en la resonancia magnética cerebral que reveló atrofia cortical generalizada y atrofia hipocampal bilateral.
Los estudios electroencefalográficos han documentado actividad ictal temporal bilateral independiente que se correlaciona clínicamente con los eventos de bradicardia e hipoxemia. Las evaluaciones neuropsicológicas seriales han demostrado déficits cognitivos multifocales que persisten a pesar del manejo integral.
El esquema terapéutico actual que combina topiramato, clobazam, levetiracetam, lacosamida y dieta cetogénica no ha logrado reducir significativamente la frecuencia de las crisis epilépticas.
Sin embargo, los familiares han reportado una mejoría subjetiva en algunos aspectos cognitivos desde la implementación de la dieta cetogénica. La persistencia de crisis focales frecuentes con generalización mensual y los déficits cognitivos establecidos evidencian la gravedad del compromiso neurológico residual y el carácter refractario de esta condición.
Según los autores (Arbey Aponte, et al), este caso clínico ilustra el curso trifásico característico del síndrome FIRES. La primera fase prodrómica consistió en una enfermedad febril que precedió en ocho días al inicio de las crisis neurológicas.
La fase aguda se manifestó como un estado epiléptico superrefractario que persistió durante seis semanas a pesar del tratamiento intensivo. Finalmente, el paciente evolucionó a la fase crónica caracterizada por epilepsia farmacorresistente con deterioro cognitivo asociado.
La discordancia entre los estudios iniciales normales y los cambios atróficos posteriores en las neuroimágenes es característica de la evolución natural de este síndrome. La pobre respuesta a múltiples intervenciones terapéuticas resalta la naturaleza particularmente refractaria de esta entidad, siendo la dieta cetogénica la modalidad que mostró algún beneficio parcial en este caso específico.