La insuficiencia suprarrenal primaria ocurre cuando las glándulas suprarrenales no producen cantidades adecuadas de cortisol y aldosterona, debido a la destrucción progresiva de su corteza.

Paciente masculino de 20 años, acudió al servicio de urgencias tras experimentar un episodio de síncope al levantarse de la cama.
Durante los últimos seis meses, había presentado síntomas progresivos que incluían fatiga severa, pérdida de peso involuntaria de aproximadamente 15 kg, náuseas, dolor abdominal difuso y mareos recurrentes.
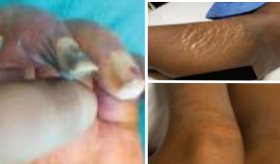
Un hallazgo físico llamativo fue la hiperpigmentación cutánea y mucosal, evidenciada por un tono bronceado en su piel y oscurecimiento de sus encías, a pesar de no haber tenido exposición solar reciente.
En la evaluación inicial, se documentó hipotensión ortostática significativa, con una presión arterial que descendió de 95/60 mmHg en posición sentada a 79/55 mmHg al ponerse de pie.
Los estudios de laboratorio revelaron anomalías electrolíticas características, incluyendo hiponatremia, hiperpotasemia e hipoglucemia, junto con una anemia leve normocrómica. Estos hallazgos, combinados con la clínica del paciente, orientaron hacia un trastorno endocrino subyacente.
El diagnóstico requirió una exclusión de otras entidades que podrían explicar esta constelación de síntomas. Según el reportaje publicado en Medscape, inicialmente se consideró hipertiroidismo, dado que podría justificar la pérdida de peso y la ansiedad referida por el paciente.
Sin embargo, este diagnóstico fue descartado ante la ausencia de taquicardia o hipertensión, sumado a un nivel normal de hormona estimulante de la tiroides (TSH).
La hemocromatosis hereditaria también formó parte del diagnóstico diferencial debido a la hiperpigmentación cutánea, pero la ferritina sérica normal y la falta de antecedentes familiares hicieron improbable esta posibilidad.
De igual manera, aunque la diabetes mellitus tipo 1 con cetoacidosis podría causar dolor abdominal y pérdida de peso, la glucemia y el pH normales eliminaron esta opción.
La combinación de síntomas constitucionales, hipotensión ortostática, hiperpigmentación y alteraciones electrolíticas (hiponatremia e hiperpotasemia) llevó a sospechar fuertemente una insuficiencia suprarrenal primaria, también conocida como enfermedad de Addison.
Esta impresión diagnóstica se confirmó mediante pruebas hormonales que mostraron un cortisol matutino bajo (130 nmol/L) y un nivel de hormona adrenocorticotrópica (ACTH) marcadamente elevado, duplicando el límite superior normal. La prueba de estimulación con ACTH sintética (cosintropina) corroboró el diagnóstico al demostrar una respuesta insuficiente de cortisol.
La insuficiencia suprarrenal primaria ocurre cuando las glándulas suprarrenales no producen cantidades adecuadas de cortisol y aldosterona, debido a la destrucción progresiva de su corteza.
En los países occidentales, aproximadamente el 90% de los casos son de origen autoinmune, donde el sistema inmunitario ataca las enzimas involucradas en la síntesis de hormonas esteroideas, particularmente la 21-hidroxilasa. Otras causas menos frecuentes incluyen infecciones (como tuberculosis o histoplasmosis), hemorragias suprarrenales, enfermedades infiltrativas (como sarcoidosis) o metástasis cancerosas.
En este paciente, la ausencia de antecedentes infecciosos o traumáticos, junto con la presencia de un trastorno autoinmune en su familia (diabetes tipo 1 en su madre), sugirieron una etiología autoinmune esporádica.
Estudios adicionales, como la detección de anticuerpos contra la 21-hidroxilasa y una tomografía computarizada de glándulas suprarrenales, pueden realizarse para confirmar la causa subyacente, aunque en este caso la presentación clínica y los hallazgos de laboratorio fueron suficientes para establecer el diagnóstico.
El abordaje terapéutico de la insuficiencia suprarrenal primaria se divide en dos fases críticas: el manejo de la crisis suprarrenal aguda y el tratamiento de mantenimiento a largo plazo.
Durante la crisis suprarrenal, caracterizada por hipotensión severa, deshidratación y alteraciones electrolíticas, el paciente requirió administración inmediata de hidrocortisona intravenosa (100 mg como dosis inicial, seguida de 200 mg en las siguientes 24 horas) junto con fluidoterapia agresiva para corregir la hipovolemia y los desequilibrios hidroelectrolíticos.
Para el manejo crónico, se inició terapia de reemplazo hormonal con glucocorticoides (hidrocortisona oral en dosis de 15-25 mg al día, fraccionada en dos o tres tomas) y mineralocorticoides (fludrocortisona en dosis de 50-300 µg diarios). La hidrocortisona suple la deficiencia de cortisol, mientras que la fludrocortisona reemplaza la aldosterona, esencial para mantener el balance de sodio, potasio y la presión arterial.
Con un tratamiento adecuado y un seguimiento endocrinológico regular, los pacientes con enfermedad de Addison pueden llevar una vida normal y activa. Sin embargo, requieren monitorización periódica para ajustar las dosis de medicación, evaluar la aparición de otras enfermedades autoinmunes asociadas (como tiroiditis o diabetes tipo 1) y prevenir complicaciones a largo plazo derivadas del exceso o déficit de reemplazo hormonal.
El caso, descrito por la Dr. Laura J. Arul, registra que tras tres meses de tratamiento, el paciente experimentó una mejoría significativa de sus síntomas, recuperó peso y pudo reintegrarse a sus actividades académicas. Su seguimiento continuará bajo supervisión especializada para garantizar un control óptimo de su condición y detectar precozmente cualquier comorbilidad asociada.