La Administración de Drogas y Alimentos de los Estados Unidos otorgó hoy la aprobación acelerada a la inyección de Vyondys 53 (golodirsen) para tratar a pacientes con distrofia muscular Duchenne (DMD) que tienen una mutación confirmada del gen de la distrofina que es susceptible a la omisión del exón 53. Se estima que alrededor del 8 por ciento de los pacientes con DMD tienen esta mutación.

Agencia Latina de Noticias Medicina y Salud Pública
La Administración de Drogas y Alimentos de los Estados Unidos otorgó hoy la aprobación acelerada a la inyección de Vyondys 53 (golodirsen) para tratar a pacientes con distrofia muscular Duchenne (DMD) que tienen una mutación confirmada del gen de la distrofina que es susceptible a la omisión del exón 53. Se estima que alrededor del 8 por ciento de los pacientes con DMD tienen esta mutación.
"La FDA reconoce la necesidad urgente de nuevos tratamientos médicos para los trastornos neurológicos graves y tenemos un compromiso de larga data de trabajar con investigadores, compañías farmacéuticas y pacientes para facilitar el desarrollo y la aprobación de tratamientos para las enfermedades raras. Con la aprobación acelerada de hoy, los pacientes con distrofia muscular Duchenne (DMD) - una enfermedad rara y devastadora - que tienen una mutación confirmada del gen de la distrofina susceptible de omisión del exón 53 ahora tendrán disponible el primer tratamiento dirigido específicamente para este subtipo de enfermedad",
dijo Billy Dunn, M.D., director interino de la Oficina de Neurociencia en el Centro de Evaluación e Investigación de Medicamentos de la FDA.
"El uso de la vía de aprobación acelerada hará que Vyondys 53 esté disponible para los pacientes sobre la base de los datos iniciales y esperamos saber más acerca de los beneficios clínicos del fármaco a partir del ensayo clínico de confirmación en curso",
agregó.
La DMD es un trastorno genético poco frecuente que se caracteriza por el deterioro y la debilidad muscular progresiva. Es el tipo más común de distrofia muscular. La DMD es causada por la ausencia de distrofina, una proteína que ayuda a mantener intactas las células musculares. Los primeros síntomas generalmente se observan entre los tres y cinco años de edad y empeoran con el tiempo.
La enfermedad a menudo ocurre en personas sin antecedentes familiares conocidos de la afección y afecta principalmente a los niños, pero en casos raros puede afectar a las niñas. La DMD ocurre en aproximadamente uno de cada 3.600 bebés varones en todo el mundo.
Las personas con DMD pierden progresivamente la capacidad de realizar actividades de forma independiente y a menudo necesitan una silla de ruedas a principios de la adolescencia. A medida que la enfermedad progresa, se pueden presentar afecciones cardíacas y respiratorias que ponen en peligro la vida. Los pacientes generalmente sucumben a la enfermedad entre los 20 y 30 años de edad; sin embargo, la gravedad de la enfermedad y la esperanza de vida varían.
Vyondys 53 fue aprobado bajo la vía de aprobación acelerada, que prevé la aprobación de medicamentos que tratan enfermedades graves o que ponen en peligro la vida y que en general ofrecen una ventaja significativa sobre los tratamientos existentes. La aprobación bajo esta vía puede basarse en estudios adecuados y bien controlados que demuestren que el medicamento tiene un efecto sobre un criterio de valoración alternativo que es razonablemente probable que prediga el beneficio clínico para los pacientes (es decir, cómo se sienten o funcionan los pacientes o si sobreviven). Esta vía proporciona a los pacientes un acceso más temprano a nuevos y prometedores medicamentos mientras la compañía lleva a cabo ensayos clínicos para verificar el beneficio clínico previsto.
La aprobación acelerada de Vyondys 53 se basa en el punto final sustituto de un aumento en la producción de distrofina en el músculo esquelético observado en algunos pacientes tratados con el fármaco. La FDA ha concluido que los datos presentados por el solicitante demostraron un aumento en la producción de distrofina que es razonablemente probable que prediga un beneficio clínico en pacientes con DMD que tienen una mutación confirmada del gen de la distrofina susceptible de omisión del exón 53. No se ha establecido un beneficio clínico del fármaco, incluida la mejora de la función motora. Al tomar esta decisión, la FDA consideró los riesgos potenciales asociados con el medicamento, la naturaleza debilitante y potencialmente mortal de la enfermedad y la falta de terapia disponible.
Vyondys 53 fue evaluado en un estudio clínico de dos partes. La primera parte incluyó 12 pacientes con DMD, ocho de los cuales recibieron Vyondys 53 y cuatro recibieron placebo. La segunda parte del estudio fue abierta e incluyó a los 12 pacientes inscritos en la primera parte del estudio y a 13 pacientes adicionales que no habían recibido previamente el tratamiento. En el estudio, los niveles de distrofina aumentaron, en promedio, de 0.10% de lo normal al inicio del estudio a 1.02% de lo normal después de 48 semanas de tratamiento con el medicamento o más.
Como parte del proceso de aprobación acelerado, la FDA está exigiendo a la compañía que realice un ensayo clínico para confirmar el beneficio clínico del medicamento. El estudio en curso está diseñado para evaluar si Vyondys 53 mejora la función motora de los pacientes con DMD con una mutación confirmada del gen de la distrofina susceptible de omisión del exón 53. Si el ensayo no verifica el beneficio clínico, la FDA puede iniciar procedimientos para retirar la aprobación del medicamento.
Los efectos secundarios más comunes reportados por los participantes que recibieron Vyondys 53 en estudios clínicos fueron dolor de cabeza, fiebre (pirexia), tos, vómitos, dolor abdominal, síntomas de resfriado (nasofaringitis) y náuseas. Reacciones de hipersensibilidad, incluyendo erupción, fiebre, picazón, urticaria, irritación de la piel (dermatitis) y exfoliación de la piel (exfoliación), han ocurrido en pacientes que fueron tratados con Vyondys 53.
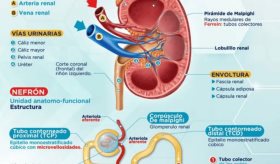
Además, se observó toxicidad renal en animales que recibieron golodirsen. Aunque no se observó toxicidad renal en los estudios clínicos con Vyondys 53, se ha observado toxicidad renal, incluyendo glomerulonefritis potencialmente mortal, después de la administración de algunos oligonucleótidos antisentido. La función renal debe ser monitoreada en pacientes que toman Vyondys 53.
La FDA otorgó a esta solicitud las designaciones de Vía Rápida y Revisión de Prioridades. Vyondys 53 también recibió la designación de Medicamento Huérfano, que proporciona incentivos para ayudar y fomentar el desarrollo de medicamentos para enfermedades raras. Además, el fabricante recibió un vale de revisión prioritaria de enfermedades pediátricas raras. El programa de vales de revisión prioritaria de enfermedades pediátricas raras de la FDA tiene la intención de fomentar el desarrollo de nuevos medicamentos y productos biológicos para prevenir y tratar las enfermedades raras en los niños.
La aprobación de Vyondys 53 fue otorgada a Sarepta Therapeutics de Cambridge, Massachusetts.