Una enfermedad hereditaria conocida en Europa, pero que es poco común también ha presentado casos en Puerto Rico. Se trara de la enfermedad de Gaucher.

Jean Mitchelle Vélez
Agencia Latina de Noticias de Medicina y Salud Pública
Una enfermedad conocida en Europa, pero que es poco común -la mayoría de pacientes hacen parte de la población judía- también ha presentado casos en Puerto Rico. Se trara de la enfermedad de Gaucher. El doctor Alain Estévez Cabrera, residente del
, presentó un caso clínico en el que el paciente comenzó a manifestar síntomas de esta condición a los
61 años
de edad. La enfermedad de Gaucher pertenece a un grupo de enfermedades lisosomales, en la que se presenta la deficiencia de una enzima encargada del procesamiento de diferentes tipos de nutrientes que ingresan al organismo, ubicada dentro de los lisosomas, uno de los organelos celulares. Hasta el momento, la comunidad médica reconoce tres tipos diferentes de la enfermedad de Gaucher. El tipo 1 es la más común y se ve en un paciente de cada
65 mil.
El tipo 2 es la más letal y se ve un caso de cada
175 mil personas.
El tipo 3, aunque similar al tipo 2 no es tan letal y se presenta en
1
de cada
200 mil
la tiene. Los síntomas que desarrolla el paciente puede variar de acuerdo al grupo que manifieste. Generalmente, los indicios de enfermedad de Gaucher son;
y bazo aumentados de tamaño y retraso mental. El tratamiento inmediato es el reemplazo de la enzima deficiente en el organismo.
En todos los tipos de enfermedad de Gaucher, el trastorno está en una mutación que existe en el cromosoma uno, que es donde se codifica la enzima cerebrosidasa. Esta encima es la encargada de metabolizar una cantidad de lípidos que se encuentran en las células, lo metaboliza, lo excretan y sigue el mecanismo de vida celular. En el caso de estos pacientes donde la enzima está ausente [...] los lípidos no se metabolizan (procesan) y se acumulan afectando la vida de la célula y llevando a la muerte tempranamente", explicó Estévez Cabrera.
En el caso clínico que se menciona, el paciente llevaba ocho años tratando
crónica sin obtener resultados. Tras estos resultados, los médicos proceden a realizar una serie de exámenes hematológicos con resultados positivos para pancitopenia. Posteriormente, realizaron exámenes de
que revelaron la presencia de algunas células conocidas como pseudo Gaucher. Los médicos inicialmente no sospecharon de la enfermedad de Gaucher por la rareza del trastorno. Por ende, se decide tratar la pancitopenia con otras alternativas con buenos resultados durante tres años. No obstante, el paciente regresa con los síntomas relacionados a la
y los médicos vuelven a hacer un chequeo para descartar alguna malignidad. En aquella oportunidad, los galenos optaron por realizar una biopsia para detectar malignidades con resultados negativos. Sin embargo, descubrieron que las células de Gaucher habían ocupado la
de un
70%
a un
80%
. Luego de este hallazgo, los tratantes deciden estudiar genéticamente al paciente para saber si se trata de un caso de enfermedad de Gaucher, diagnóstico que en ese momento se descartó ya que los análisis arrojaron resultados negativos para esta condición. La genetista encargada de los resultados, sospechó de una mutación. Como paso siguiente se estudió el
exhaustivamente, dando como respuesta ser un afectado por la enfermedad de Gaucher tipo 1. Ya en esta etapa, el paciente estaba en condiciones de salud precarias, donde requería transfusiones de sangre seguidas cada dos semanas y desarrolló otras complicaciones. Una situación que se logró trabajar para que sus transfusiones fueran en periodos más largos de tiempo (cada 2 meses), mejorando notablemente la salud del afectado.
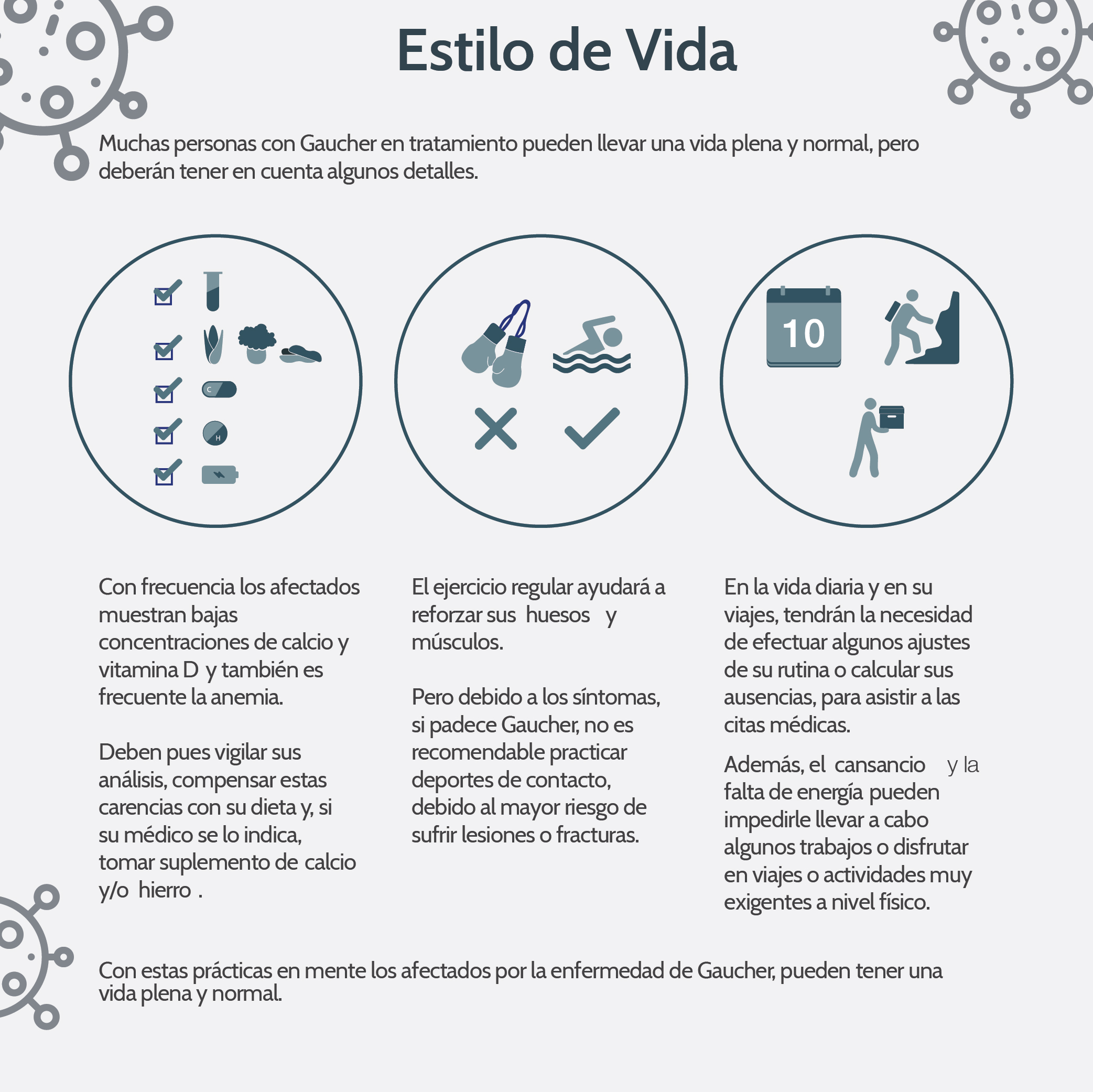
En el tipo 1, como este caso, (los pacientes) llegan a edades adultas, ya que la mutación no es total y no produce ausencia de la enzima, sino que hay un funcionamiento parcial o anormal. Estos pacientes pueden llegar a una edad adulta asintomáticos, aunque es una enfermedad bastante progresiva. Tenía 61 años cuando es remitido por primera vez a hematología. Es una enfermedad genética pero se trató por todos los medios para que la familia pudiese ser sometida a pruebas, pero todos los miembros se negaron", agregó el médico, quien es de ascendencia cubana.
Se cree que la familia negó realizarse los exámenes ya que el paciente tenía un grado de retraso mental acelerado y sus hermanos no mostraron ningún tipo de interés para conocer la mutación, o, si en efecto alguno también padecía de la rara enfermedad. Con el tiempo la enfermedad fue deteriorando el sistema hematológico y el paciente murió a causa de una neumonía. Pero cabe destacar que tuvo una supervivencia de 10 años luego de recibir el diagnóstico.