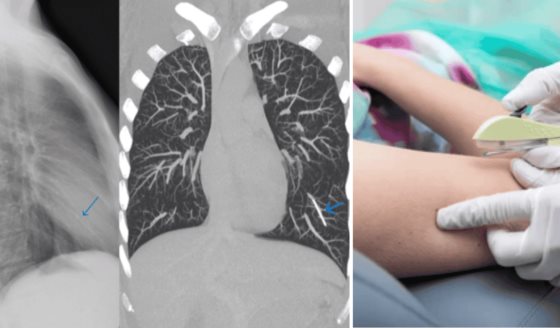
Después del nacimiento, la prueba de Silverman-Anderson reveló una puntuación de 0, indicando una adaptación respiratoria inicial adecuada.

Se presenta el caso de un bebé de 48 horas de nacido, ingresado en el servicio de urgencias pediátricas tras presentar múltiples síntomas entre ellos la dificultad respiratoria, anomalías corporales, entre otros.
Durante el embarazo, la madre recibió atención prenatal desde el segundo mes de gestación, refiriendo el consumo de ácido fólico y sulfato ferroso. Sin embargo, presentó enfermedad inflamatoria pélvica, tratada con supositorios vaginales. En la última ecografía gestacional se reportó macrocefalia fetal.
El bebé nació por retraso del crecimiento intrauterino, la puntuación de Apgar fue de 8 al primer minuto y de 9 a los cinco minutos. La prueba de Silverman-Anderson reveló una puntuación de 0, indicando una adaptación respiratoria inicial adecuada. Sin embargo, el recién nacido presentó una succión débil y, debido a limitaciones en los recursos del hospital donde nació, fue remitido a un hospital de más alto nivel.
A su ingreso, el neonato fue sometido a ventilación con presión positiva continua debido a dificultad respiratoria. Durante la exploración física se detectó un soplo holosistólico grado 2 en la zona de auscultación de la válvula mitral.
Una radiografía de tórax evidenció un vértice cardiaco elevado hacia la izquierda, arteria pulmonar excavada, dilatación severa de la aurícula derecha y disminución de la vasculatura pulmonar. Además, el electrocardiograma mostró ritmo sinusal con desviación del eje derecho y onda R alta en V1.
La macrocefalia, acompañada de fontanelas amplias (anterior de 6x6 cm y posterior de 2x2 cm), llevó a realizar una tomografía computarizada (TC) cerebral, la cual reveló agenesia del cuerpo calloso, hidrocefalia con dilatación del cuarto ventrículo e hipoplasia del tentorio cerebeloso. Con base en estos hallazgos, se consultó al departamento de neurocirugía, que indicó una derivación ventriculoperitoneal para el manejo de la hidrocefalia.
Al cuarto día de hospitalización, un ecocardiograma convencional reveló levocardia, hipertrofia ventricular izquierda, comunicación interventricular de 10 mm con un shunt mixto izquierda-derecha, y un pequeño conducto arterioso persistente. Estos hallazgos respaldaron el diagnóstico de tetralogía de Fallot (TOF), siendo este un hallazgo inusual dentro del síndrome agenesia del cuerpo calloso y otras anomalías sistémicas (ACOGS por sus siglas en inglés .
El neonato presentaba múltiples rasgos dismórficos, incluyendo frente prominente, orejas de implantación baja, ojos hundidos con entropión, labios finos y fisuras palpebrales bajas. También se observaron hipoplasia de los músculos rectos abdominales y criptorquidia bilateral. Estas características inicialmente sugirieron un posible síndrome del abdomen en ciruela pasa, pero dicha hipótesis fue descartada al identificar TOF y agenesia del cuerpo calloso.
La identificación de múltiples malformaciones mayores y menores permitió descartar cromosomopatías como trisomía 13, 18 y 21. Tras una exhaustiva revisión bibliográfica, se estableció el diagnóstico de ACOGS, un síndrome descrito por primera vez en 2019 y asociado a mutaciones en el gen CDH2 del cromosoma 18q12, el cual codifica la proteína N-cadherina.
Los autores del caso (Montero-Vázquez et al) refieren que, el recién nacido permaneció hospitalizado durante 10 días, tiempo durante el cual se le retiró la oxigenoterapia y se estabilizó su condición. Debido a las limitaciones del hospital en términos de recursos para manejo especializado, fue referido a un hospital pediátrico de alta complejidad para el tratamiento quirúrgico integral de la cardiopatía y la hidrocefalia, además de establecer un abordaje multidisciplinario.
El ACOGS es un síndrome del neurodesarrollo caracterizado por retraso global del desarrollo, agenesia o hipoplasia del cuerpo calloso y anomalías craneofaciales, cardiacas, oculares y genitales. Este caso reporta el primer diagnóstico de TOF asociado al síndrome, ampliando el espectro fenotípico conocido.
La mutación del gen CDH2 afecta la adhesión y migración celular, explicando las malformaciones observadas. Las N-cadherinas desempeñan un papel crucial en el desarrollo neural, la sinaptogénesis y la formación del tubo neural, así como en la función cardíaca y el desarrollo ocular.